
Synergistic Combination of KRAS Small Molecule Inhibitors and Mutation-Specific mRNA Vaccines: A Novel Paradigm for KRAS-Mutant Cancers

Received: 10 March 2026 |
Revised: 5 April 2026 |
Accepted: 15 April 2026
Abstract
KRAS is one of the most frequently mutated oncogenes in human cancers and was long considered "undruggable." Although the successful development of small molecule inhibitors targeting the KRAS G12C mutation, such as Sotorasib and Adagrasib, represents a historic breakthrough in targeted therapy, their clinical application is hampered by significant challenges, including limited target coverage, modest objective response rates, and the inevitable development of acquired resistance. Concurrently, the success of mRNA vaccine technology in infectious disease prevention has validated its potential as a powerful platform, offering a new paradigm for cancer therapy. By presenting mutated KRAS as a neoantigen, mRNA cancer vaccines can elicit a specific T-cell mediated immune response to precisely eliminate tumor cells. This approach holds the unique advantages of covering a broader range of KRAS mutations (e.g., G12D, G12V) and inducing long-lasting immunological memory.
This article systematically elucidates an innovative "targeted therapy plus immunotherapy" combination strategy, integrating KRAS small molecule inhibitors with mutation-specific KRAS mRNA vaccines. The central thesis is that these two modalities can exert synergistic anti-tumor effects through complementary mechanisms. On one hand, KRAS inhibitors not only directly suppress tumor growth but may also foster a "hot tumor" microenvironment more permissive to T-cell infiltration by inducing immunogenic cell death and reversing immunosuppression. On the other hand, the mRNA vaccine can precisely eradicate tumor cells that have developed resistance to inhibitors via mechanisms such as bypass pathway activation, as well as clear minimal residual disease, thereby overcoming drug resistance and preventing tumor recurrence. By reviewing the mechanisms of action, clinical progress, and limitations of each therapy, this paper provides an in-depth analysis of the theoretical underpinnings for their synergistic combination, offering a strong rationale and a forward-looking perspective for the development of a more effective and durable treatment paradigm for KRAS-mutant cancers.
Keywords: KRAS; mRNA vaccine; nucleic acid therapy; combination therapy; tumor immune microenvironment; resistance mechanisms
1 Introduction
1.1 Research Background
The KRAS gene is the most frequently mutated oncogene in human cancers, acting as a central hub in signaling pathways that regulate critical cellular processes such as proliferation, survival, and metabolism [1][2]. KRAS mutations are highly prevalent in several aggressive malignancies, occurring in over 90% of pancreatic ductal adenocarcinomas (PDAC), approximately 40-50% of colorectal cancers (CRC), and around 25-30% of non-small cell lung cancers (NSCLC) [2][3]. For nearly four decades, KRAS was deemed an "undruggable" target, posing a formidable challenge to drug development. This was due to its smooth surface topology, which lacks conventional binding pockets for small molecules, and its extremely high affinity for GTP [4][5].
In recent years, however, a deeper understanding of the allosteric states of the KRAS protein has led to the successful development of covalent inhibitors specifically targeting the KRAS G12C mutation, marking a historic breakthrough in KRAS-targeted therapy [4]. The advent of these small-molecule drugs successfully rendered the KRAS target "druggable" for the first time, ushering in a new era of precision medicine for KRAS-mutant cancers.
Concurrently, mRNA technology has robustly demonstrated its tremendous potential as a vaccine platform during the global fight against the COVID-19 pandemic, distinguished by its rapid development speed and high protective efficacy [6][7]. This success has significantly propelled the application of mRNA technology into therapeutic fields, especially fueling a surge in the research and development of personalized cancer vaccines. It signals the maturation of a novel strategy to combat cancer by precisely modulating the body's immune system [8][9].
1.2 KRAS Therapy: From Monotherapy to Combination Strategies
Sotorasib (AMG 510) and Adagrasib (MRTX849), as approved KRAS G12C inhibitors, have brought significant survival benefits to patients with advanced cancers harboring the KRAS G12C mutation, such as NSCLC, achieving objective response rates (ORR) of 30-40% [4][10]. Despite this initial progress, the long-term efficacy of these small-molecule inhibitors is severely limited by drug resistance [11]. The core challenges are twofold: first, currently approved drugs are only effective against the KRAS G12C mutation and are ineffective against other codon 12 mutations (e.g., G12D, G12V) that are more prevalent in pancreatic and colorectal cancers [3][12]; second, nearly all treated patients eventually develop acquired resistance [4][11]. The mechanisms of this resistance are highly complex, primarily involving secondary mutations in the KRAS protein itself or the activation of KRAS-independent bypass signaling pathways [13][14].
The therapeutic strategy of mRNA cancer vaccines represents a fundamental shift. Instead of directly interfering with the biochemical function of the KRAS protein, it utilizes the KRAS mutation site as a highly specific tumor neoantigen. Through vaccination, this neoantigen is presented to the immune system, thereby training and activating T cells that can precisely recognize and eliminate tumor cells expressing this "mutational marker" [6][7].
Through precise sequence design, mRNA vaccines can be engineered to target any KRAS mutation type (G12C, G12D, G12V, etc.), and it is even possible to create "multivalent vaccines" containing multiple mutations to achieve truly personalized therapy [15]. Since the immune system targets the tumor cells expressing the KRAS mutant antigen itself, rather than inhibiting their aberrant signaling activity, these cells remain susceptible to immune attack as long as they continue to present the mutant KRAS antigen, even if they have developed resistance to small-molecule inhibitors through bypass pathway activation [16].
1.3 Research Aim
Given that KRAS small-molecule inhibitors have demonstrated limitations in clinical application, such as narrow target coverage and the propensity for resistance, while mRNA vaccines offer a complementary therapeutic effect through a distinct mechanism of action, the central aim of this paper is to propose and systematically explore the synergistic mechanisms of combining KRAS small-molecule inhibitors with mutation-specific KRAS mRNA vaccines. This study analyzes how this "targeted therapy + immunotherapy" combination strategy can achieve a synergistic ("1+1>2") therapeutic effect. The goal is to provide a novel and innovative therapeutic paradigm with high clinical translational potential for overcoming resistance to KRAS-targeted therapy and enhancing clinical outcomes.
2 KRAS-Targeted Therapeutic Strategies
2.1 KRAS Small-Molecule Inhibitors
Directly targeting the KRAS protein, particularly with covalent inhibitors designed to exploit its mutation site, represents a major breakthrough in recent anti-tumor drug development. These drugs function by irreversibly binding to the KRAS protein, locking it in an inactive state and thereby directly blocking its oncogenic signal transduction.
2.1.1 Mechanism of Action and Clinical Achievements
The core mechanism of first-generation KRAS G12C inhibitors, represented by Sotorasib (AMG 510) and Adagrasib (MRTX849), is their irreversible covalent binding. The KRAS protein cycles between an active GTP-bound state (KRAS-ON) and an inactive GDP-bound state (KRAS-OFF) (Figure 1). The G12C mutation introduces a cysteine residue that, in the KRAS-OFF state, is exposed within a specific binding pocket known as the Switch-II pocket. Sotorasib and Adagrasib specifically recognize and form a covalent bond with the thiol group of this cysteine residue, thereby stabilizing KRAS in its inactive, GDP-bound conformation. This locking effect prevents KRAS from interacting with its downstream effector proteins (such as RAF and PI3K), effectively inhibiting the constitutive activation of critical oncogenic signaling pathways like the MAPK pathway [4][17].
The clinical value of these inhibitors has been validated in several pivotal trials. In the Phase II CodeBreaK-100 trial, Sotorasib treatment for patients with previously treated KRAS G12C-mutant non-small cell lung cancer (NSCLC) resulted in an objective response rate (ORR) of 37.1%, a median progression-free survival (PFS) of 6.8 months, and a median overall survival (OS) of 12.5 months, leading to its accelerated approval by the U.S. FDA. Similarly, in the KRYSTAL-1 trial, Adagrasib demonstrated significant efficacy in pre-treated KRAS G12C-mutant NSCLC patients, with an ORR of 42.9%, a median PFS of 6.5 months, and a median OS of 12.6 months. Notably, Adagrasib also showed encouraging intracranial activity in patients with central nervous system (CNS) metastases. These achievements mark the first time that KRAS was successfully converted from an "undruggable" to a "druggable" target, providing a new therapeutic option for a specific patient population [4][17].
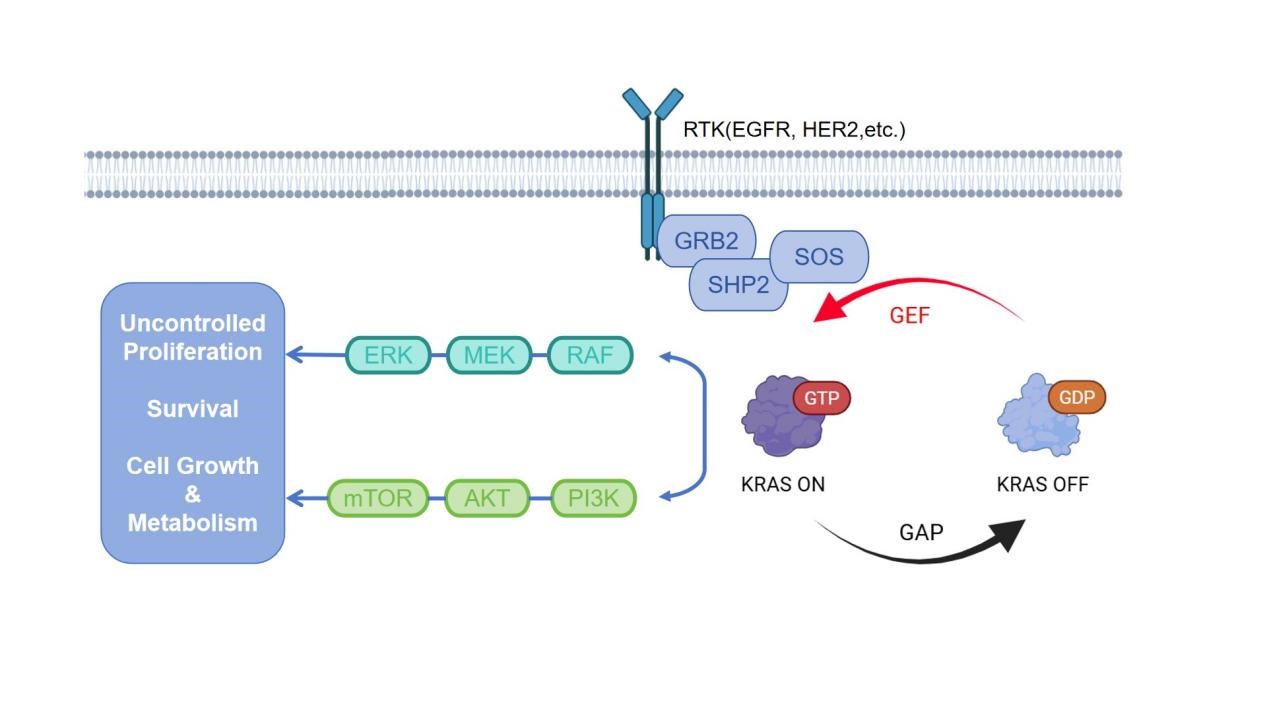
Figure 1. Schematic diagram of the KRAS signaling pathway and its downstream effector pathways (adapted from Prior et al. [1]).
2.1.2 Resistance Mechanisms
Although KRAS G12C inhibitors have shown initial clinical success, acquired resistance remains a major challenge, similar to other targeted therapies. Systematic analyses have revealed that resistance mechanisms can be broadly categorized into two types: on-target resistance and bypass resistance.
On-target resistance refers to drug failure caused by alterations in the KRAS gene itself, manifesting as secondary mutations and KRAS gene amplification. Secondary mutations occur under the selective pressure of the drug, leading to new mutations in the KRAS gene. These mutations may either directly alter the G12C site or occur near the drug-binding pocket, sterically hindering drug binding. KRAS gene amplification, wherein tumor cells increase the copy number of the KRAS G12C allele, reduces the effective intracellular drug concentration, thereby diminishing the inhibitory effect [4][18].
Bypass resistance, which is more common in clinical practice, occurs when tumor cells activate alternative signaling pathways to circumvent their dependency on KRAS G12C, thereby reactivating downstream signaling, such as the MAPK pathway. Specific mechanisms include: the activation or amplification of receptor tyrosine kinases (RTKs) such as EGFR, MET, or FGFR; the activation of other RAS isoforms like NRAS or HRAS, which drives the reactivation of upstream signals in pathways like MAPK or PI3K/AKT; mutations or amplifications in downstream molecules such as BRAF, MEK, or PIK3CA, rendering signal transduction independent of the upstream KRAS node; and histological transformation, such as the transition of non-small cell lung cancer to squamous cell carcinoma or small cell lung cancer, which then rely on KRAS-independent survival pathways [13][14].
2.2 mRNA Immunotherapy
Unlike small-molecule drugs that directly target oncoproteins, mRNA cancer vaccines aim to mobilize the patient's own immune system, enabling it to specifically recognize and eliminate tumor cells, representing a highly personalized and dynamic therapeutic strategy.
2.2.1 Mechanism of Action
First, tumor-specific neoantigens, including those arising from KRAS mutations, are identified by sequencing the patient's tumor tissue. The mRNA sequences encoding these neoantigens are then synthesized and encapsulated in a highly efficient delivery vehicle, such as lipid nanoparticles (LNPs) [7][15]. Following vaccination, the LNPs are efficiently taken up by antigen-presenting cells (APCs), particularly dendritic cells (DCs). Once inside the cytoplasm, the mRNA is translated into mutated protein fragments by the cell's translational machinery [6][7]. Subsequently, these neoantigen peptides are degraded by the proteasome within the APCs and presented on the cell surface via both Major Histocompatibility Complex (MHC) Class I and Class II molecules. The neoantigen-loaded APCs then migrate to the lymph nodes, where they present the antigens to CD8+ and CD4+ T cells, respectively. This process activates cytotoxic T lymphocytes (CD8+ T cells) and helper T cells (CD4+ T cells) that specifically recognize the neoantigen, leading to the generation of effector and memory T cells capable of specifically identifying and eliminating tumor cells that express the mutated protein, thereby achieving systemic immune surveillance and killing [6][7].
2.2.2 Clinical Advantages
The design of mRNA vaccines relies solely on the genetic sequence, not the three-dimensional protein structure. Theoretically, this allows them to target any mutation, including the highly prevalent G12D and G12V mutations for which no effective small-molecule inhibitors are currently available [15]. Successful vaccination can induce the formation of long-lasting memory T cells, providing the body with continuous tumor immune surveillance. This is crucial for preventing recurrence and metastasis and represents a unique advantage not offered by small-molecule drugs [15].
The potential of personalized mRNA neoantigen vaccines has been preliminarily validated in clinical settings. The individualized neoantigen vaccine mRNA-4157 (V940), co-developed by Moderna and Merck, although not specifically designed for KRAS, has validated the feasibility of this platform. In the Phase IIb KEYNOTE-942 trial for the adjuvant treatment of high-risk melanoma, mRNA-4157 in combination with the PD-1 inhibitor pembrolizumab significantly reduced the risk of recurrence or death by 44% compared to pembrolizumab monotherapy, and it received Breakthrough Therapy Designation from the FDA. Autogene cevumeran (BNT122), an individualized neoantigen vaccine co-developed by BioNTech and Genentech (a member of the Roche Group), has also shown positive signals in early-phase clinical trials. A study in post-operative pancreatic ductal adenocarcinoma patients found that the vaccine induced a robust T-cell response specifically targeting neoantigens, including KRAS, in approximately 50% of patients. Patients who mounted an immune response demonstrated a longer recurrence-free survival [19][20].
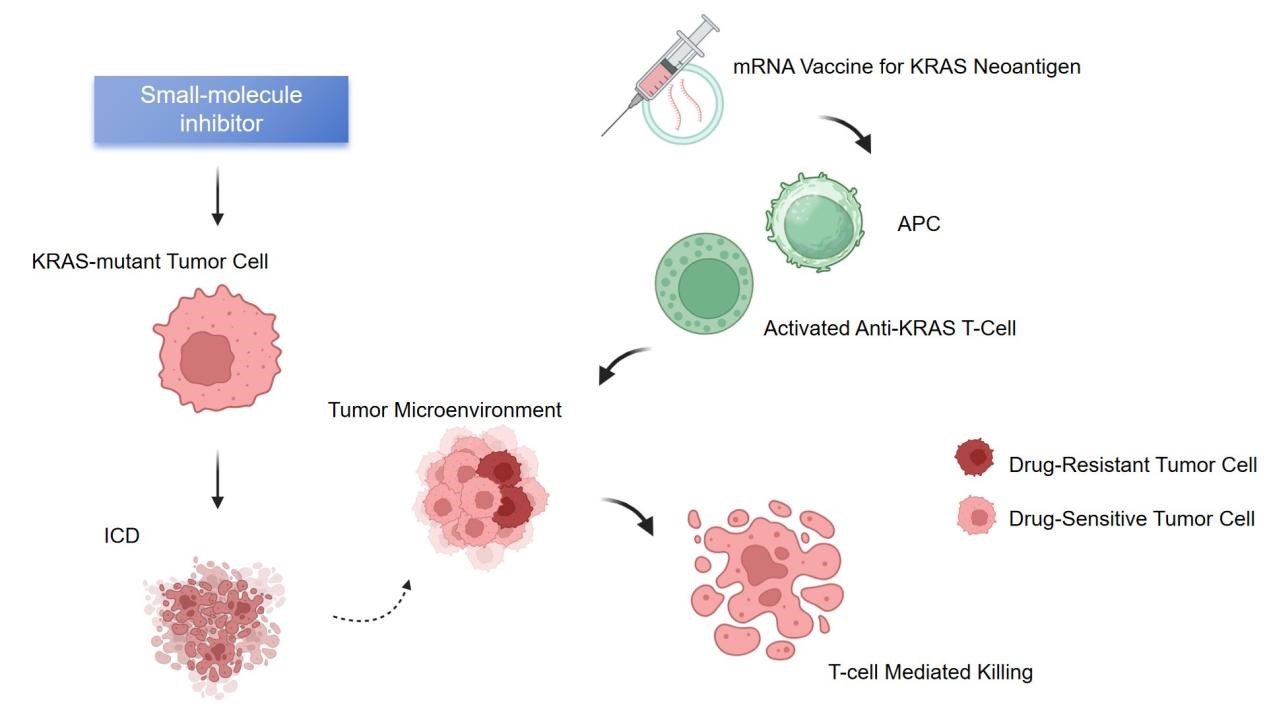
Figure 2. The synergistic mechanism of combining KRAS-targeted therapy and immunotherapy.
2.3 Rationale for Combination Therapy
KRAS small-molecule inhibitors and mRNA vaccines are perfectly complementary in terms of their mechanisms of action, advantages, and limitations (Figure 2). Therefore, combining these two modalities is expected to achieve synergistic efficacy, representing a significant future direction for KRAS-targeted therapy.
2.3.1 Reshaping the Immune Microenvironment
KRAS inhibitors not only directly suppress tumor cell proliferation but also reshape the tumor microenvironment (TME) through multiple pathways, converting it from an immunosuppressive "cold" tumor into a "hot" tumor that is responsive to immune cell attack. This creates a favorable environment for the mRNA vaccine to exert its effects.
On one hand, some KRAS inhibitors, while inducing tumor cell death, can trigger a specific form of cell death known as Immunogenic Cell Death (ICD). This process is accompanied by the release of various "danger signals" or Damage-Associated Molecular Patterns (DAMPs), such as surface-exposed calreticulin (CRT), extracellularly released adenosine triphosphate (ATP), and high mobility group box 1 (HMGB1) [21][22]. These DAMPs can strongly recruit and promote the maturation of antigen-presenting cells, such as dendritic cells, to the tumor site. This effectively creates an "endogenous vaccine effect" in situ, which can greatly enhance the specific immune response subsequently activated by the mRNA vaccine [21][23].
On the other hand, a constitutively active KRAS oncogenic pathway actively fosters an immunosuppressive TME in various ways, such as by driving tumor cells to secrete immunosuppressive cytokines like transforming growth factor-β (TGF-β) and interleukin-10 (IL-10), and by recruiting immunosuppressive cells like regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs) to infiltrate the TME [24][25]. The use of KRAS inhibitors can directly block this constitutive activation of the KRAS pathway, thereby reducing the secretion of immunosuppressive cytokines and the infiltration of immunosuppressive cells. Concurrently, this may increase the recruitment of cytotoxic CD8+ T cells to the tumor site and enhance their function, thus removing immunological barriers for the effector T cells activated by the mRNA vaccine, allowing them to more effectively penetrate the tumor tissue and execute their killing function [5][26].
2.3.2 Overcoming Resistance Mechanisms
Conversely, the mRNA vaccine can precisely eliminate resistant cells and minimal residual disease that emerge after treatment with small-molecule inhibitors.
Eliminating resistant cells is one of the core rationales for this combination therapy. When tumor cells develop resistance to KRAS inhibitors through mechanisms like bypass activation, they are no longer sensitive to the small-molecule drug. However, these resistant cells typically still express the mutated KRAS protein that serves as their hallmark. Therefore, for T cells that have been activated by an mRNA vaccine to target the KRAS mutation, these resistant cells remain clearly identifiable targets that can be specifically recognized and eliminated. This orthogonal killing mechanism significantly reduces the probability of the tumor developing simultaneous resistance to both therapies [16].
After treatment with a small-molecule inhibitor, undetectable minimal residual disease (MRD) or dormant tumor cells may persist, even if imaging studies show tumor disappearance. The long-term immunological memory system induced by the mRNA vaccine can provide continuous surveillance for months or even years post-treatment. If these residual cells attempt to proliferate, memory T cells can be rapidly activated to eliminate them in their nascent stages, leading to a deeper and more durable complete response and potentially preventing disease recurrence. Furthermore, the initial T-cell attack triggered by the vaccine, upon killing tumor cells, releases a broader array of tumor-associated antigens (not limited to the KRAS mutation). This may activate a more extensive T-cell response against these new antigens, creating a self-reinforcing anti-cancer immune cycle and further broadening the breadth and depth of the anti-tumor immunity [16].
3 Clinical Translational Challenges and Counterstrategies for Combination Therapy
Although the combination strategy of KRAS small molecule inhibitors and mRNA vaccines shows significant theoretical advantages, a series of key challenges must be overcome to transition from the laboratory to clinical application. These challenges span multiple dimensions, including vaccine preparation, treatment regimen optimization, and inter-patient variability. Only by proposing targeted solutions can the successful translation of this combination therapy be advanced.
3.1 Technical Bottlenecks in Vaccine Preparation and Delivery
The efficiency of mRNA vaccine preparation and in vivo delivery represents a core technical bottleneck constraining its clinical application. On one hand, the preparation of personalized mRNA vaccines relies on precise sequencing and analysis of a patient's tumor mutation profile. Currently, the cost of whole-exome sequencing is high and the turnaround time is long (typically 2-4 weeks) [27]. However, patients with advanced cancer often experience rapid disease progression and cannot afford such a lengthy waiting period for vaccine manufacturing. On the other hand, although lipid nanoparticles (LNPs) are the most widely used mRNA delivery vector at present, they still suffer from insufficient targeting. While LNPs are efficiently taken up by antigen-presenting cells, they are also susceptible to non-specific clearance by Kupffer cells in the liver [28]. This reduces the efficiency of vaccine enrichment in crucial sites such as tumor-draining lymph nodes, thereby compromising the strength of the immune response [29].
To address the aforementioned issues, optimization can be pursued from two directions. First is the development of a hybrid vaccine strategy that combines "rapid customization with universal coverage." For high-frequency KRAS mutations (e.g., G12C, G12D, G12V), standardized "off-the-shelf" mRNA vaccine modules could be prepared in advance. If these common mutations are detected after patient sequencing, these universal modules can be directly combined with patient-specific modules for low-frequency mutations, significantly shortening the preparation time [30]. The second approach involves designing more targeted delivery vectors. For instance, modifying the LNP surface with dendritic cell-specific antibodies (e.g., anti-CD11c) or chemokine receptor ligands (e.g., CCL21) can guide LNPs to precisely target dendritic cells in tumor-draining lymph nodes. This would reduce non-specific uptake by the liver and enhance the bioavailability of the vaccine.
3.2 Challenges in Optimizing Treatment Regimens
The administration sequence, dosage ratio, and treatment duration of the combination therapy directly impact the synergistic effect, and currently, there is no unified standard for optimization. For example, administering a high-dose KRAS inhibitor first may rapidly reduce tumor volume, but it could also lead to massive tumor cell death, releasing excessive "interfering signals" that, in turn, suppress the antigen-presenting function of dendritic cells [31]. Conversely, if the mRNA vaccine is administered first, the T cells it activates may be unable to effectively infiltrate the tumor tissue if the tumor microenvironment remains in an immunosuppressive state [32]. Furthermore, significant variations exist among patients in terms of their tolerance to inhibitors and the intensity of their immune response to vaccines, rendering a "one-size-fits-all" treatment regimen inadequate for meeting individualized needs.
To solve this problem, an optimal regimen can be explored by integrating preclinical models with clinical studies. In the preclinical stage, Patient-Derived Xenograft (PDX) models harboring different KRAS mutations can be used to test various administration sequences, such as "low-dose inhibitor pretreatment → vaccination → maintenance-dose inhibitor" and "vaccination → inhibitor therapy." By assessing indicators like the infiltration of CD8+ T cells and the proportion of immunosuppressive cells within the tumor microenvironment, the most synergistic regimen can be identified [33]. In the clinical stage, an "adaptive clinical trial design" can be employed. This approach allows for real-time adjustments of the inhibitor dosage and vaccination interval based on dynamic patient efficacy (e.g., tumor shrinkage rate) and safety metrics (e.g., incidence of immune-related adverse events), thereby achieving "dynamic and personalized" treatment.
3.3 Obstacles from Tumor Microenvironment Heterogeneity
Even if the combination therapy can effectively activate T cells, the heterogeneity of the tumor microenvironment (TME) may still render some tumor regions as "immune-privileged zones," preventing T-cell infiltration and function. For instance, solid tumors like pancreatic ductal adenocarcinoma often feature a dense fibrotic stroma that acts as a physical barrier to T-cell infiltration [34]. Some tumor cells may also downregulate the expression of MHC molecules to evade T-cell recognition [35]. Moreover, metabolic abnormalities within the TME, such as high lactate levels and hypoxia, can inhibit the proliferation and cytotoxic functions of T cells [36].
To address this issue, incorporating a "TME modulator" into the combination therapy to form a "triple-therapy" strategy can be considered. First, agents such as matrix metalloproteinase inhibitors (e.g., Marimastat) can be used to degrade the tumor's fibrotic barrier and promote T-cell infiltration [37]. Second, co-administration of epigenetic drugs (e.g., HDAC inhibitors) can upregulate MHC molecule expression on the surface of tumor cells, enhancing their recognition by T cells [38]. Third, correcting metabolic abnormalities by inhibiting lactate dehydrogenase (e.g., GNE-140) or ameliorating the hypoxic environment (e.g., using hypoxia-activated hemoglobin carriers) can restore the cytotoxic function of T cells [39].
4 Future Outlook
With continuous breakthroughs in KRAS-targeted therapies and mRNA vaccine technologies, their combination strategy is poised to achieve significant progress in the following three directions, bringing new hope to the treatment of KRAS-mutant cancers.
1) Multi-target "Pan-KRAS" Combination Therapy
Current combination strategies primarily target a single KRAS mutation subtype. In the future, this could be expanded into a "pan-KRAS" combination therapy. On one hand, this involves developing broad-spectrum inhibitors that can simultaneously target multiple KRAS mutations (e.g., G12C/G12D/G12V). For example, the recently developed KRAS G12D inhibitor, MRTX1133, has demonstrated potent inhibitory effects on G12D-mutant tumors in preclinical models [40]. On the other hand, it entails designing "multivalent mRNA vaccines" that incorporate antigens from various KRAS mutations. These vaccines could even include co-mutation antigens associated with the KRAS pathway (e.g., TP53, PIK3CA mutations) to activate a broader T-cell response, cover the heterogeneous mutations within the tumor, and further reduce the risk of drug resistance.
2) Synergistic Expansion with Immune Checkpoint Inhibitors
Combining KRAS inhibitors, mRNA vaccines, and immune checkpoint inhibitors (ICIs) (e.g., PD-1/PD-L1 inhibitors) to form a "triple therapy" may produce an even stronger synergistic effect. T cells activated by the mRNA vaccine may become "exhausted" upon entering the TME due to PD-L1 expression on tumor cells. A PD-1 inhibitor can lift this immunosuppression and restore T-cell cytotoxicity. Meanwhile, the KRAS inhibitor reshapes the microenvironment, further enhancing T-cell infiltration. Preclinical studies have already shown that combining a KRAS G12C inhibitor with a PD-1 inhibitor significantly extends the survival of mouse models [41]. Adding an mRNA vaccine to this regimen holds the potential to further improve efficacy. Future clinical trials will be needed to validate the safety and effectiveness of this "triple therapy" in advanced KRAS-mutant cancers.
3) Application in Adjuvant Therapy for Early-Stage Cancer
While current research on combination therapy focuses mainly on advanced cancers, its application could be extended to the adjuvant setting for early-stage disease. For patients with early-stage KRAS-mutant cancers who have undergone surgical resection (e.g., Stage I non-small cell lung cancer, Stage II colorectal cancer), a postoperative combination of a low-dose KRAS inhibitor and an mRNA vaccine could potentially eliminate micrometastatic disease, establish long-lasting immune memory, and thereby lower the risk of recurrence. For example, in the adjuvant setting for pancreatic cancer, the 5-year survival rate with current regimens is less than 20%. Adjuvant therapy with an mRNA vaccine and inhibitor combination could significantly improve long-term survival by enhancing immune surveillance [30]. The value of this combination strategy in the adjuvant treatment of early-stage cancer will need to be validated through large-scale Phase III clinical trials [42].
5 Conclusion
As the most prevalent oncogenic driver in human cancers, KRAS mutations have witnessed a historic shift from "undruggable" to "precision-targeted" therapies. However, the limitations of small-molecule inhibitor targets and the issue of drug resistance remain fundamentally unresolved. The emergence of mRNA cancer vaccine technology offers a novel immunological perspective for treating KRAS-mutated cancers. By targeting KRAS-mutant neoantigens to activate specific T-cell responses, this approach not only covers multiple KRAS mutation subtypes but also induces long-lasting immune memory, forming a perfect mechanism complement to small-molecule inhibitors.
This paper systematically elucidates the synergistic mechanism of combining KRAS small-molecule inhibitors with mRNA vaccines: Inhibitors create an "attackable" environment for vaccine-activated T cells by inducing immunogenic cell death and reversing the immunosuppressive microenvironment. while vaccines eliminate drug-resistant cells and minimal residual disease, overcoming inhibitor resistance. Together, they achieve the dual objectives of tumor shrinkage and relapse prevention. Although this combination strategy faces challenges in clinical translation---including vaccine preparation, regimen optimization, and microenvironment heterogeneity---these obstacles may be progressively addressed through "universal + personalized" vaccine design, adaptive clinical protocols, and the introduction of microenvironment modulators.
Looking ahead, with ongoing exploration of pan-KRAS therapies, triple combination regimens, and early adjuvant treatments, the combination strategy of KRAS small-molecule inhibitors and mRNA vaccines will continue to evolve. It holds promise as a new paradigm for treating KRAS-mutated cancers, offering hope for long-term survival to more patients.
References
[1] Prior IA, Hood FE, Hartley JL. The frequency of Ras mutations in cancer. Cancer Res. 2020;80(14):2969-2974.
[2] Uniyal P, et al. KRAS mutations in cancer: understanding signaling pathways to immune regulation and the potential of immunotherapy. Cancers. 2025;17(5):785.
[3] Yang Y, et al. KRAS mutations in solid tumors: characteristics, current therapeutic strategy, and potential treatment exploration. J Clin Med. 2023;12(2):709.
[4] Chour A, et al. Mechanisms of resistance to KRASG12C inhibitors in NSCLC. Front Oncol. 2024;14:1328728.
[5] Orlen M, et al. T-cell dependency of tumor regressions with RAS (ON) multi-selective inhibition. Cancer Discov. 2025;15(8):1697-1716.
[6] Laila UE, Wang A, Xu ZX. Emerging prospects of mRNA cancer vaccines. Front Immunol. 2024;15:1448489.
[7] Ni L. Advances in mRNA-based cancer vaccines. Vaccines. 2023;11(10):1599.
[8] Wang B, et al. Recent advances in mRNA cancer vaccines: meeting challenges and embracing opportunities. Front Immunol. 2023;14:1246682.
[9] Biernat AK, et al. A Review of mRNA Vaccines in Prostate and Lung Cancer Therapy. Int J Innov Technol Soc Sci. 2025;1(3):1-15.
[10] Oya Y, Mitsudomi T. Is adagrasib just another sotorasib? Transl Lung Cancer Res. 2023;12(5):940.
[11] Désage AL, et al. Targeting KRAS Mutant in Non-Small Cell Lung Cancer. Front Oncol. 2022;12:796832.
[12] Rubinson DA, et al. Sotorasib Is a Pan-RAS G12C Inhibitor Capable of Driving Clinical Response in NRAS G12C Cancers. Cancer Discov. 2024;14(5):727-736.
[13] Dunnett-Kane V, et al. Mechanisms of resistance to KRASG12C inhibitors. Cancers. 2021;13(1):151.
[14] Lv X, et al. Modulation of the proteostasis network promotes tumor resistance to oncogenic KRAS inhibitors. Science. 2023;381(6662):1065.
[15] Vishweshwaraiah YL, Dokholyan NV. mRNA vaccines for cancer immunotherapy. Front Immunol. 2022;13:1029069.
[16] Miyashita H, Kato S, Hong DS. KRAS G12C inhibitor combination therapies. Front Oncol. 2024;14:1380584.
[17] Peng TR, et al. Comparative efficacy of adagrasib and sotorasib in KRAS G12C-mutant NSCLC. Cancers. 2024;16(21):3676.
[18] Awad MM, et al. Acquired resistance to KRASG12C inhibition in cancer. N Engl J Med. 2021;384(25):2382-2393.
[19] Khattak A, et al. Distant metastasis-free survival results from the randomized, phase 2 mRNA-4157-P201/KEYNOTE-942 trial. J Clin Oncol. 2023;41(17):LBA9503.
[20] Weber JS, et al. Individualised neoantigen therapy mRNA-4157 (V940) plus pembrolizumab versus pembrolizumab monotherapy in resected melanoma (KEYNOTE-942): a randomised, phase 2b study. Lancet. 2024;403(10427):632-644.
[21] Troitskaya OS, et al. Immunogenic Cell Death in Cancer Therapy. Acta Naturae. 2022;14(1):40-53.
[22] Arimoto KI, et al. Emerging role of immunogenic cell death in cancer immunotherapy. Front Immunol. 2024;15:1390263.
[23] Kroemer G, et al. Immunogenic cell death in cancer therapy. Annu Rev Immunol. 2013;31:51-72.
[24] Xu M, et al. Unveiling the role of KRAS in tumor immune microenvironment. Biomed Pharmacother. 2024;171:116058.
[25] Cullis J, et al. Kras and Tumor Immunity: Friend or Foe? Cold Spring Harb Perspect Med. 2018;8(9):a031849.
[26] Uniyal P, et al. KRAS mutations in cancer: understanding signaling pathways to immune regulation and the potential of immunotherapy. Cancers. 2025;17(5):785.
[27] Sahin U, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 2017;547(7662):222-226.
[28] Akinc A, et al. The Onpattro story and the clinical translation of nanomedicines containing nucleic acid-based drugs. Nat Nanotechnol. 2019;14(12):1084-1087.
[29] Lokugamage MP, et al. Optimization of lipid nanoparticles for the delivery of nebulized therapeutic mRNA to the lungs. Nat Biomed Eng. 2021;5(9):1059-1068.
[30] Rojas LA, et al. Personalized RNA neoantigen vaccines stimulate T cells in pancreatic cancer. Nature. 2023;618(7963):144-150.
[31] Canon J, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. 2019;575(7781):217-223.
[32] Liu J, et al. Cancer vaccines as promising immuno-therapeutics: platforms and current progress. J Hematol Oncol. 2022;15(1):1-26.
[33] Jenkins RW, et al. Ex Vivo Profiling of PD-1 Blockade Using Organotypic Tumor Spheroids. Cancer Discov. 2018;8(2):196-215.
[34] Jiang H, et al. Tumor-associated fibrosis as a regulator of tumor immunity and response to immunotherapy. Cancer Immunol Immunother. 2017;66(8):1037-1048.
[35] Cornel AM, et al. MHC Class I Downregulation in Cancer: Underlying Mechanisms and Potential Targets for Cancer Immunotherapy. Cancers. 2020;12(7):1760.
[36] Fischer K, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood. 2007;109(9):3812-3819.
[37] Hua H, et al. Matrix metalloproteinases in tumorigenesis: an evolving paradigm. Cell Mol Life Sci. 2011;68(23):3853-3868.
[38] Emanuele S, et al. Histone deacetylase inhibitors: apoptotic effects and clinical implications (Review). Int J Oncol. 2008;33(4):637-646.
[39] Chen H, et al. Metabolic heterogeneity and immunocompetence of infiltrating immune cells in the breast cancer microenvironment (Review). Oncol Rep. 2021;45(3):846-856.
[40] Hallin J, et al. The KRASG12C Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020;10(1):54-71.
[41] Canon J, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. 2019;575(7781):217-223.
[42] Kotecha R, et al. Adagrasib in Non-Small-Cell Lung Cancer. N Engl J Med. 2022;387(13):1238-1239.

